On April 8th, 2024, a total solar eclipse will sweep across North America, from Mexico to the Maine-Canadian border. For those who experienced the spectacular solar eclipse of 2017, this one will be similar, crossing the United States from west to east and passing...
In the summer of 2017, my brother Geoffrey landed in the Northwestern Hospital emergency room confused, with a high-grade fever, and so dizzy he couldn’t walk. He had a severe lung infection and he could barely breathe – a pulmonary function test revealed that his lung function had dropped to 23%.
After doctors stabilized him in the ER, they admitted him as an inpatient, and he was soon visited by the nurse practitioner who has overseen his care for the past 18 years.
“How the hell did you get to 23% lung function?” she exclaimed as she walked through the door. Typically, their interactions had been friendly, sarcastic, and cordial — she’s like a family member to us — but this time, she was mad at Geoffrey and she was getting nervous. Mirroring what we had been picking up as a family, she and the rest of Geoffrey’s pulmonary team had seen his numbers drop more quickly over the last few years as he had developed allergies to some of the antibiotics that keep his chronic lung infections in check.
Since he was about 17, Geoffrey normally spent two weeks out of every year as an inpatient for a “tune-up,” as we like to call it, where he received respiratory therapy twice a day and a daily course of IV antibiotics to combat his chronic lung infections. But over the last few years, those stays became longer, sometimes creeping up to three weeks, and more frequently, reaching almost twice a year.
So when his nurse saw him enter Northwestern through the ER with a lung function of 23%, that was her last straw. His doctors came in and told him that he had to start thinking about a lung transplant.
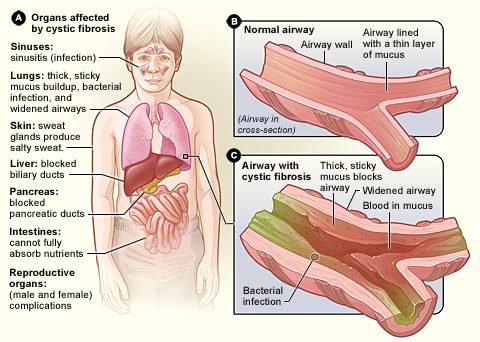
Geoffrey had cystic fibrosis, the most common lethal genetic disease in the United States. It causes a person’s internal organs, from the lungs to the pancreas to the intestines, to get covered in a thick coat of gross, sticky mucus that damages tissue and clogs ducts.
When Geoffrey was born, CF was called a childhood disease because most patients did not survive past fifth grade. Now, the life expectancy for someone born today with CF is about 44 years old. For a CF patient, the key to living the longest, fullest life possible is keeping the lungs healthy.
CF is so destructive to the body because it causes an infrastructure problem: the body’s major passageway for salt is broken. This passageway, or channel, normally allows chloride ions to cross over from one side of your cell membranes to the other. They do this because, like us, chloride ions like to remain socially distanced; they don’t want to have to squeeze past each other inside our cells. They’d rather spread out, and an open tunnel to the outside of the cell lets them do this.
In someone with CF like my brother, however, this passageway is missing. As a result, chloride gets stuck inside its crowded cellular home, where it really starts to ache for some personal space.
Since chloride can’t move out, the only way to give it the room it wants is to dilute it by bringing water in. But when water rushes inside his cells, it leaves a sticky mess behind. The dried-out mucus clogs ducts, irritates the lining of organs, and most importantly, attracts lethal bacterial infections that thrive in such a gross environment.
The mucus kills the delicate structures in the lungs that help them absorb oxygen and release carbon dioxide, which means my brother never got the same amount of oxygen in a breath that a healthy person would. The mucus in his body also damaged his pancreas, the organ that makes the enzymes that help us digest our food and releases the hormones that regulate our blood sugar, including insulin. As a result, Geoffrey had to take enzymes (in pills) with every meal and constantly keep an eye on his blood sugar in case it got too high or dropped too low. The hardest part of CF-related diabetes, as this is called, is its unpredictability. After mucus hurts the pancreas in a CF patient, it still works sometimes, meaning it might release a bit of insulin once in a while, causing sudden drops in blood sugar.
In addition to the digestive enzymes and insulin, Geoffrey took a lot of other medications to keep himself healthy. He took antibiotics all the time to keep his chronic lung infections in check, he inhaled medications that dilute the mucus in his lungs to help wash it away, and he took various supplements to keep his other organ systems, especially his digestive system, in working order.
To make things worse, with all of these medications comes another complication: kidney damage. Over the years, antibiotics can damage the kidneys and reduce their ability to filter toxins out of the body.
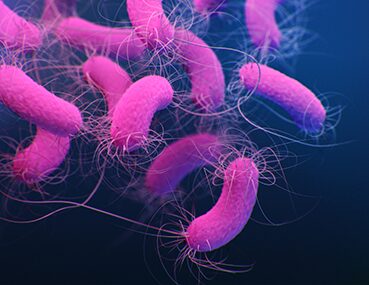
While all of these problems can be scary, it’s the chronic lung infections that are the most threatening. Like most patients with CF, Geoffrey harbored an infection caused by a specific species of bacteria called P. aerogenosa (air-oh-jen-OH-suh). This bacteria lives in all of us, but in patients with CF, the environment in the lungs makes it easier for them to thrive.
It’s because of these bacteria that most CF patients become candidates for lung transplants. An immensely risky procedure, it’s the last resort for getting rid of the nasty lung infections that threaten lives. The new lungs don’t have CF, making them less susceptible to infection. But since they come from a donor, the recipient must take medication that suppresses their immune systems for the rest of their lives and must always worry about the looming threat of infections and organ rejection. Geoffrey lost this battle earlier this year.
Hopefully, most CF patients in the future can wait a lot longer before they even have to think about organ transplantation. In 2019, the FDA approved the first treatment that targets the underlying cause of cystic fibrosis. It’s called Trikafta and many consider it a miracle drug because instead of just treating the symptoms of CF, the drug cocktail actually rebuilds and opens the salt bridges that CF patients are missing. This treatment is supposed to work in 90% of CF patients, and hopefully, it’ll lead to even longer lives. Meanwhile, researchers are also looking at ways to get around antibiotic resistance and treat CF patients’ lethal lung infections using viruses. But that technology is still under development.
In the meantime, there are still 30,000 patients with CF in the US who need our help — and we still need a treatment for those remaining 10% of patients who don’t have any good options. Visit cff.org to learn more about what you can do to help.